Gaussian RMSd
The standard alignment process in RMSd is optimal in the sense that minimizes the variance between ensembles of structures, but presents some limitations when applied to anisomorphic and flexible molecules such as proteins, where we
can observe flexible regions like loops or hinges which generate large and localized movements in specific regions of the proteins.
The standard alignment process distribute the variance in all residues, providing a more complex to understand flexibility picture (see Figure 1 and Damm and Carlson 2006).

Figure 1. Detail of the movement of a small domain with respect to the large one and the associated variance plot. Left as determined by an alignment where the big domain is fully aligned in each structure of the ensemble, Right as determined from a normal RMSd-like alignment process.
In a situation like that shown in Figure 1, an human expert will perform the alignment considering only the large domain, focusing then all the structural variability into the small one.
Unfortunately, without human intervention, decision of the number of residues to be considered in the alignment is more difficult.
In our server we implement an iterative algorithm based on the procedure used in the robust linear regression.
Accordingly, we defined a weighted RMSd (eq. 2), where  is a weight factor ranging from 0 to 1 which modulates the impact in the rRMSd of the
is a weight factor ranging from 0 to 1 which modulates the impact in the rRMSd of the 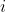 residue/atom (note that the equation converges to normal RMSd for all weight factors equal to 1)
residue/atom (note that the equation converges to normal RMSd for all weight factors equal to 1)
where:
The weight factor is determined with a gaussian term based on the distance between each residue/atom pair, assigning high weightings (∼1) to static domains and lower weightings to more flexible regions like loops or hinges (∼0) (eq. 4)
We have chosen a value of 2 Ų for the arbitrary scaling factor c (Damm and Carlson 2006), taking into account that the structures that we are comparing are very similar (standard RMSd < 5Å) as we are working with molecular dynamics snapshots of the same structure.
Note that equations 2 and 4 are interdependent via the alignment and accordingly need to be solved iteratively (default maximum number iterations 99) until convergence in gRMSd.
A residue in a flexible loop will have originally a weight of one, leading to an illogical alignment.
However, eq. 4 will detect that even with the global alignment the residue is quite mobile and will reduce its weight in the gRMSd function, reducing in parallel its weight into the new alignment.
Subsequent cycles will maximize this effect, leading to an overall alignment constructed considering as much more relevant the rigid blocks of the protein.