Input Data
FlexServ works on protein structures. Only Cα atoms are considered for simulation. Structures with more than one chain of missing residues are acceptable, but be aware that the obtained results may not be accurate as the methods used are usually calibrated with single chain molecules.
The following types of input data are accepted:
-
Protein structure in PDB format.
-
PDB Code. The structure corresponding to the entered code is retrieved from the Protein Data Bank. Optionally, a specific chain, or combination of chains, can be requested.
-
Protein sequence. It can be inserted using FASTA format or specified using a valid UniprotKB code. BLAST is used to search the sequence in a non-redundant database of valid PDB entries. An E-value limit of 1.10-5 is used to assure that the retrieved structure is a valid homologue of the sequence provided. Please note that only a limited number of UniprotKB sequences have homologues in the Protein Data Bank.
-
User provided trajectory. User can provide his/her own trajectory using the PCAZIP compressed format. Tools to manage pcazip formats can be downloaded
here. The length of the trajectory is limited to 1000 snapshots.
-
MoDEL trajectories. Pre-calculated atomístic MD trajectories, obtained within the
MoDEL project are also available. They can be selected using their PDB id.
-
Reload analysis. Data obtained from a previous FlexServ run can be reloaded to repeat the analysis.
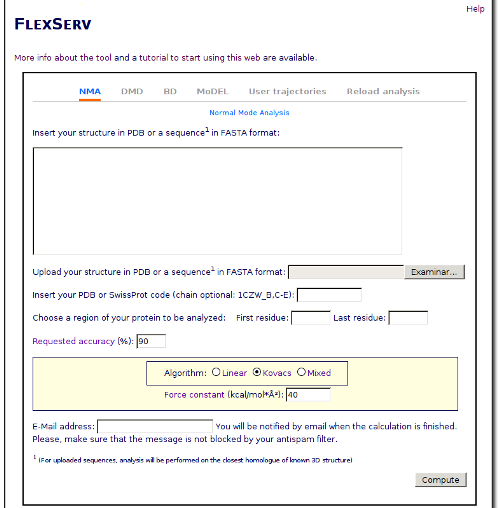
According to the selected type of simulation, a number of parameters are necessary. Default values for a typical run are already provided.
Parameters
-
Requested accurancy: Percentage of the variance that will be taken into account when selecting how many eigenvectors include in the compressed trajectory. Higher values give a better fidelity to the original trajectory, but lead to poorer compression factors. The default value used in FlexServ is 90%.
- Normal Mode Analysis (NMA)
- Algorithm: Choose the algorithm to use: Linear, Kovacs, or Mixed.
- Force Constant: Measure of the strength of the spring connecting atoms measured in Kcal/mol·Å²
- Cut-off radius: Maximum distance for the pairs of atoms to be included in the calculation in Å.
- Discrete Dynamics (DMD)
- Cut-off radius: Maximum distance for the pairs of atoms to be included in the calculation in Å.
- Temperature: Used to define velocities of simulated particles.
- Sigma: Well amplitude for consecutive Cα atoms
- Sigma Go: Well amplitude for non-consecutive Cα.
- Brownian Dynamics (BD)
- Steps: Number of time steps that will be performed during the simulation.
- dt: Time ellapsed between two consecutive time steps.
- Output frequency: Number of frames after which a frame is written and considered for analysis.
- Const: Measure of the strength of the spring connecting atoms measured in Kcal/mol·Å²
- r0: Mean distance between consecutive Cα atoms in Å.
-
User Trajectories: Users are required to upload the trajectory compressed in PCAZIP format. This file format can be processed with the
PCASuite suite of tools.
-
Reload Analysis: Users are required to upload the original FlexServ analysis package.